- New to JMP? Join us Sept. 23-24 for the Early User Edition of Discovery Summit, tailor-made for new users. Register now for free!
- Your voice matters! Tell us how you prefer to receive JMP updates, so we can tailor our communication to your needs. Take short survey.
- See how to access JMP Marketplace - and - find, create & share add-ins to extend your JMP. Watch video.
- Subscribe to RSS Feed
- Mark Topic as New
- Mark Topic as Read
- Float this Topic for Current User
- Bookmark
- Subscribe
- Mute
- Printer Friendly Page
Discussions
Solve problems, and share tips and tricks with other JMP users.- JMP User Community
- :
- Discussions
- :
- JMP and R connection issue

- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Get Direct Link
- Report Inappropriate Content
JMP and R connection issue
I just saw a cool webinar today on using some features in R that are not available in JMP via JSL. I am specifically talking about t-SNE: Data visualization with t-SNE and UMAP .
I am running JMP 14 on Mac OS 10.13 (High Sierra) and, according to the add-in link above, the R version to get is 3.3.3, so I downloaded that, restarted, and followed this: https://www.jmp.com/support/help/en/15.1/index.shtml#page/jmp/installing-r.shtml#. I cannot get even the very simple script in this trouble-shooting guide to work, not an error, just nothing. The JMP scripting guide is written for Windows, but I tried manually setting the "set environmental variable" to the applications folder, but that did not work either. R works and is stored in my Applications folder. I had older versions of R on my computer, but I deleted them (and restarted afterwards). I made it to where I can read and write files (it is my personal computer, and I am the administrator), yet even "R Init ();" does nothing.
As of a few months ago, JMP14, R ver. 3.3.3., and MAC OS10.13 were all theoretically compatible. Did anything happen in the meantime that would have made it to where I can no longer use R via the script window?

- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Get Direct Link
- Report Inappropriate Content
Re: JMP and R connection issue
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Get Direct Link
- Report Inappropriate Content
Re: JMP and R connection issue
Nay you worry, they are on the case (though thus far stumped). My goal is to, once we figure out the issue, post the solution here so that other Mac users looking to run R via JSL don't run into the same issue.

- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Get Direct Link
- Report Inappropriate Content
Re: JMP and R connection issue
@abmayfield Yeah, Mac had some versioning issues previously. One of the Mac users was able to connect to R and managed to run this Embedding add-in but it was a while ago and many factors have changed. I hope the tech support will figure it out for you.
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Get Direct Link
- Report Inappropriate Content
Re: JMP and R connection issue
I figured it out! It is actually an R vs. Mac issue.
https://community.rstudio.com/t/i-can-t-install-packages-on-r-studio-non-zero-exit-status/52135
If you try to install the Rtsne package on a Mac, R will install it, then immediately delete it and give you all kinds of weird error messages UNLESS you install it via the Mac-specific registry mentioned in this link (https://mac.R-project.org). This is the kind of thing that makes me prefer JMP over R. Even installing a simple data package with R took me 36 hours to trouble-shoot and figure out. Could t-SNE be integrated into the next version of JMP Genomics maybe?
There is a weird, but minor, JMP issue, though. "R Init" does not work at all on my Mac. Only "R is connected" will cause JMP and R to sync. From reading various posts, it seems like BOTH should be able to connect to R, so it's strange that "R Init" fails to do so. In other words, Mac users on 10.13 with R 3.3.3 will need to use "R is connected" not "R Init" to use R via JSL.
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Get Direct Link
- Report Inappropriate Content
Re: JMP and R connection issue
@abmayfield I'm glad that you figured that out. I was not aware of R connect/init issue on Mac, my Embedding add-in might not work because I use R init () to connect to R. I will update it. Regarding JMPG, we have a new Single-cell RNA-seq workflow used part of this Embedding add-in as an optional visualization method but only if you have the right versions of R and Rtsne/UMAP been installed. Also, I want to point out that JMP Genomics is only for Windows users, for now if you want a copy of it for Mac, you might need to set up some virtual Windows instance in order to use it. We are working on a native version of t-SNE for JMP, but not sure when it will be available though.
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Get Direct Link
- Report Inappropriate Content
Re: JMP and R connection issue
Actually, I can reproduce your demo t-SNE plots in JMP Pro ver. 14 with R 3.3.3 on Mac OS 10.13. The only error I get has to do with fonts, but otherwise it works.
More importantly, I actually wonder if it's suitable for my needs. I only have a few dozen samples at most and a few hundred features (i.e., proteins) at most. I'm thinking, then, that the t-SNE won't really give me a very different solution to PCA or MDS. In fact, it looks like it won't run at all: when I tried 12 samples x 94,000 genes with a perplexity of 5, it said the perplexity was still too high:
Error in Rtsne.default(inDataTsne, dims = 2, perplexity = 5, verbose = TRUE, :
Perplexity is too large.
Error in eval(expr, envir, enclos) : object 'outputY' not found
Unexpected errors occurred while attempting to transfer the data.
Issues found in R, could be caused by unsuccessful installation of Rtsne/umap packages or limited memory.
However, it also mentioned some R error, so I can't tell if it's my data's fault or R's fault!
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Get Direct Link
- Report Inappropriate Content
Re: JMP and R connection issue
@abmayfield You are absolutely right about the sample size, tSNE and UMAP are intended for larger sample size, usually at least a few hundreds. In the original paper of UMAP, it actually specifically recommended at least 500 samples for a robust result. Since you only have 12 samples but 94,000 genes, you may consider first reduce the number of genes by only select those vary between samples (variable genes), then further reduce the dimension using PCA or SVD analysis. You can view clusters with the top principal components or you can use those top principal components as features for clustering algorithms such as Hierarchical clustering in JMP, the dendrogram or constellation plot should give you a pretty decent view of clusters in your sample.
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Get Direct Link
- Report Inappropriate Content
Re: JMP and R connection issue
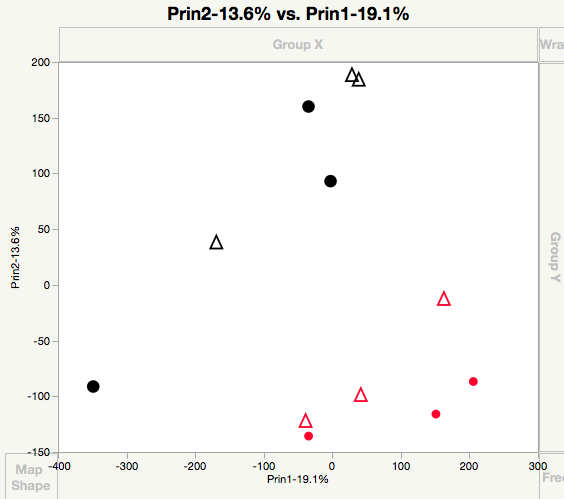
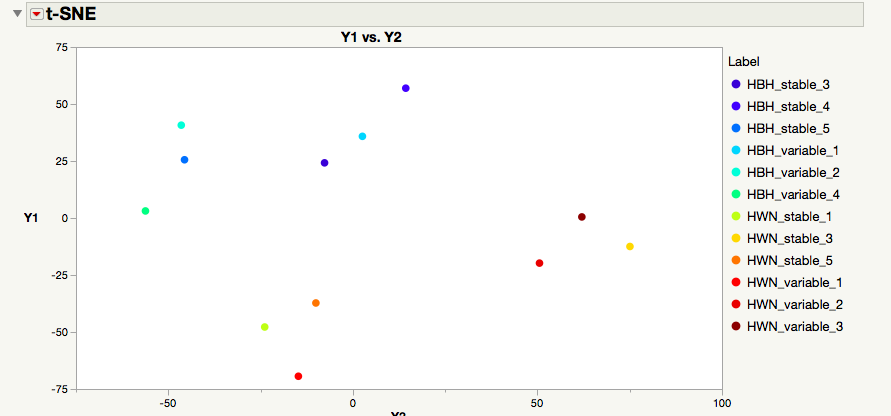
I did end up trying it with this 12 sample x 95,000 gene transcriptome after my last post, and it's interesting because I can only do 300-400 iterations or it will crash at high perplexities (anything over 5)! Maybe no surprise. But if I lower the perplexity to 3-4 and the iterations to 200-300, I get a plot that is actually pretty interesting. Corals from my two sites show some nice separation (as was evident in the PCA=first screen shot attached). See second image attached for the t-SNE (HBH and HWN are the abbreviations for the two sites).
I did notice one property of t-SNE that was noted in the original paper: unlike PCA, you don't always get the same solution twice! In fact, they can vary WILDLY. Some solutions are worthy of Nature papers whereas the next might be a low impact factor marine biology journal, haha. So I can definitely see some pros and cons of the method. But all that being said, I think you are right in that I'd need many more samples to make it practical. Anyway, I am glad to know that it works in JMP since my wife does scRNA-Seq with cell cultures, so, while it may not be the best for me at the moment, she can certainly benefit from the add-in you made!
{kind=link}
{kind=link}
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Get Direct Link
- Report Inappropriate Content
Re: JMP and R connection issue
Thank you for sharing. It was definitely a right move to lower perplexity considering the small sample size. The tSNE plot did capture your sites nicely, it seems like you have 4 local communities in your data (assume the algorithm worked correctly). I hope this finding is worthy of a Nature paper, good luck. :)
BTW, we will have materials that related to scRNA-seq added to JMP Genomics site if your wife is interested.
Recommended Articles
- © 2026 JMP Statistical Discovery LLC. All Rights Reserved.
- Terms of Use
- Privacy Statement
- Contact Us